PADCEV® DOSING OVERVIEW
Starting patients on PADCEV1
1L PADCEV + Pembrolizumab Combination Dosing
30-minute infusion time for PADCEV
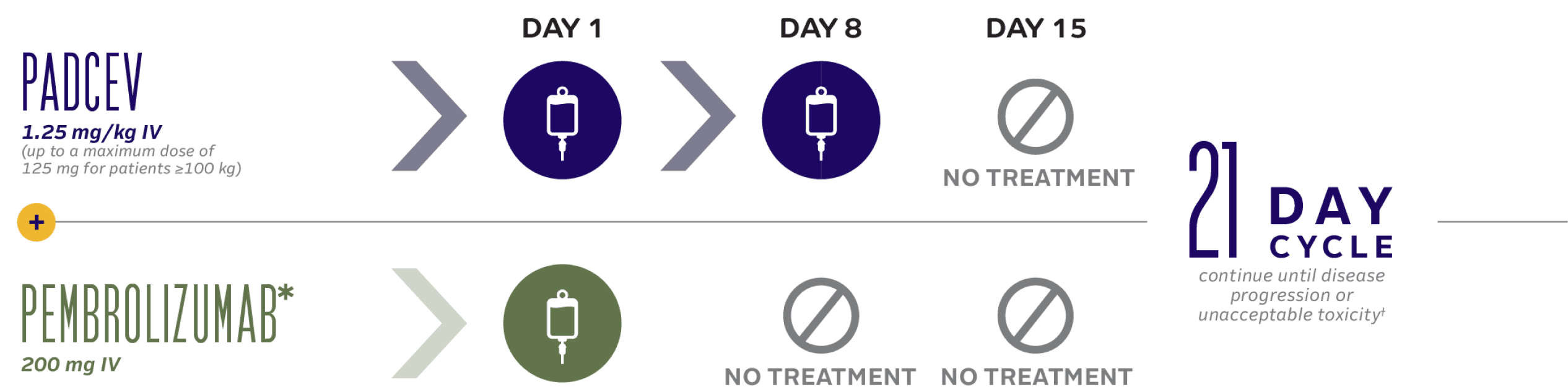

*This was the dosing schedule followed for pembrolizumab in the EV-302 clinical trial. Refer to the pembrolizumab Prescribing Information for the recommended dosing information for pembrolizumab.1
†In the absence of disease progression or unacceptable toxicity, pembrolizumab was continued for up to 2 years.1
2L+ PADCEV Monotherapy Dosing
30-minute infusion time for PADCEV

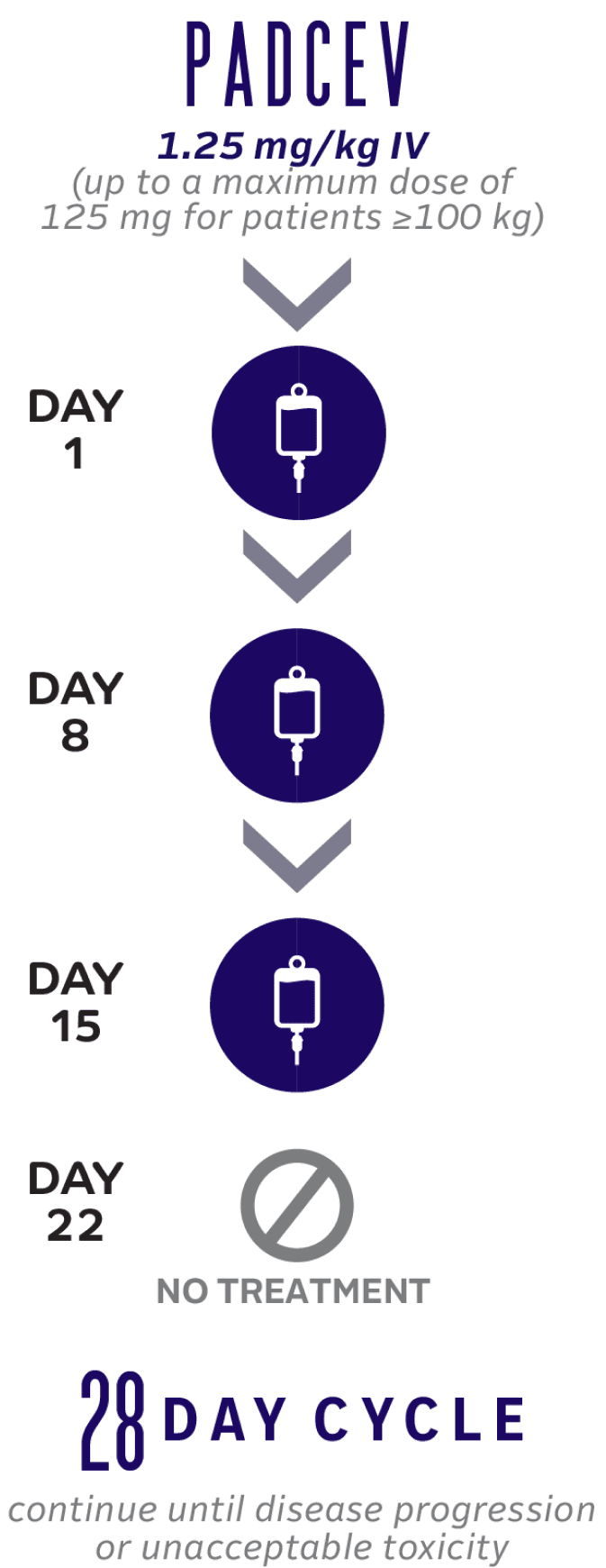
1L=first-line; 2L=second-line; IV=intravenous.
Reference: 1. PADCEV [package insert]. Northbrook, IL: Astellas Pharma US, Inc.
Important Safety Information/Indication
BOXED WARNING: SERIOUS SKIN REACTIONS
• PADCEV® can cause severe and fatal cutaneous adverse reactions including Stevens-Johnson syndrome (SJS) and Toxic Epidermal Necrolysis (TEN), which occurred predominantly during the first cycle of treatment, but may occur later.
• Closely monitor patients for skin reactions.
• Immediately withhold PADCEV and consider referral for specialized care for suspected SJS or TEN or severe skin reactions.
• Permanently discontinue PADCEV in patients with confirmed SJS or TEN; or Grade 4 or recurrent Grade 3 skin reactions.
INDICATION
PADCEV, in combination with pembrolizumab, is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC).
PADCEV, as a single agent, is indicated for the treatment of adult patients with locally advanced or mUC who:
• have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and platinum-containing chemotherapy, or
• are ineligible for cisplatin-containing chemotherapy and have previously received one or more prior lines of therapy.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Skin reactions Severe cutaneous adverse reactions, including fatal cases of SJS or TEN occurred in patients treated with PADCEV. SJS and TEN occurred predominantly during the first cycle of treatment but may occur later. Skin reactions occurred in 70% (all grades) of the 564 patients treated with PADCEV in combination with pembrolizumab in clinical trials. When PADCEV was given in combination with pembrolizumab, the incidence of skin reactions, including severe events, occurred at a higher rate compared to PADCEV as a single agent. The majority of the skin reactions that occurred with combination therapy included maculo-papular rash, macular rash and papular rash. Grade 3-4 skin reactions occurred in 17% of patients (Grade 3: 16%, Grade 4: 1%), including maculo-papular rash, bullous dermatitis, dermatitis, exfoliative dermatitis, pemphigoid, rash, erythematous rash, macular rash, and papular rash. A fatal reaction of bullous dermatitis occurred in one patient (0.2%). The median time to onset of severe skin reactions was 1.7 months (range: 0.1 to 17.2 months). Skin reactions led to discontinuation of PADCEV in 6% of patients.
Skin reactions occurred in 58% (all grades) of the 720 patients treated with PADCEV as a single agent in clinical trials. Twenty-three percent (23%) of patients had maculo-papular rash and 34% had pruritus. Grade 3-4 skin reactions occurred in 14% of patients, including maculo-papular rash, erythematous rash, rash or drug eruption, symmetrical drug-related intertriginous and flexural exanthema (SDRIFE), bullous dermatitis, exfoliative dermatitis, and palmar-plantar erythrodysesthesia. The median time to onset of severe skin reactions was 0.6 months (range: 0.1 to 8 months). Among patients experiencing a skin reaction leading to dose interruption who then restarted PADCEV (n=75), 24% of patients restarting at the same dose and 24% of patients restarting at a reduced dose experienced recurrent severe skin reactions. Skin reactions led to discontinuation of PADCEV in 3.1% of patients.
Monitor patients closely throughout treatment for skin reactions. Consider topical corticosteroids and antihistamines, as clinically indicated. For persistent or recurrent Grade 2 skin reactions, consider withholding PADCEV until Grade ≤1. Withhold PADCEV and refer for specialized care for suspected SJS, TEN or for Grade 3 skin reactions. Permanently discontinue PADCEV in patients with confirmed SJS or TEN; or Grade 4 or recurrent Grade 3 skin reactions.
Hyperglycemia and diabetic ketoacidosis (DKA), including fatal events, occurred in patients with and without pre-existing diabetes mellitus, treated with PADCEV. Patients with baseline hemoglobin A1C ≥8% were excluded from clinical trials. In clinical trials of PADCEV as a single agent, 17% of the 720 patients treated with PADCEV developed hyperglycemia of any grade; 7% of patients developed Grade 3-4 hyperglycemia (Grade 3: 6.5%, Grade 4: 0.6%). Fatal events of hyperglycemia and DKA occurred in one patient each (0.1%). The incidence of Grade 3-4 hyperglycemia increased consistently in patients with higher body mass index and in patients with higher baseline A1C. The median time to onset of hyperglycemia was 0.5 months (range: 0 to 20 months). Hyperglycemia led to discontinuation of PADCEV in 0.7% of patients. Five percent (5%) of patients required initiation of insulin therapy for treatment of hyperglycemia. Of the patients who initiated insulin therapy for treatment of hyperglycemia, 66% (23/35) discontinued insulin at the time of last evaluation. Closely monitor blood glucose levels in patients with, or at risk for, diabetes mellitus or hyperglycemia. If blood glucose is elevated (>250 mg/dL), withhold PADCEV.
Pneumonitis/Interstitial Lung Disease (ILD) Severe, life-threatening or fatal pneumonitis/ILD occurred in patients treated with PADCEV. When PADCEV was given in combination with pembrolizumab, 10% of the 564 patients treated with combination therapy had pneumonitis/ILD of any grade and 4% had Grade 3-4. A fatal event of pneumonitis/ILD occurred in two patients (0.4%). The incidence of pneumonitis/ILD, including severe events, occurred at a higher rate when PADCEV was given in combination with pembrolizumab compared to PADCEV as a single agent. The median time to onset of any grade pneumonitis/ILD was 4 months (range: 0.3 to 26 months).
In clinical trials of PADCEV as a single agent, 3% of the 720 patients treated with PADCEV had pneumonitis/ILD of any grade and 0.8% had Grade 3-4. The median time to onset of any grade pneumonitis/ILD was 2.9 months (range: 0.6 to 6 months).
Monitor patients for signs and symptoms indicative of pneumonitis/ILD such as hypoxia, cough, dyspnea or interstitial infiltrates on radiologic exams. Evaluate and exclude infectious, neoplastic and other causes for such signs and symptoms through appropriate investigations. Withhold PADCEV for patients who develop Grade 2 pneumonitis/ILD and consider dose reduction. Permanently discontinue PADCEV in all patients with Grade 3 or 4 pneumonitis/ILD.
Peripheral neuropathy (PN) When PADCEV was given in combination with pembrolizumab, 67% of the 564 patients treated with combination therapy had PN of any grade, 36% had Grade 2 neuropathy, and 7% had Grade 3 neuropathy. The incidence of PN occurred at a higher rate when PADCEV was given in combination with pembrolizumab compared to PADCEV as a single agent. The median time to onset of Grade ≥2 PN was 6 months (range: 0.3 to 25 months).
PN occurred in 53% of the 720 patients treated with PADCEV as a single agent in clinical trials including 38% with sensory neuropathy, 8% with muscular weakness and 7% with motor neuropathy. Thirty percent of patients experienced Grade 2 reactions and 5% experienced Grade 3-4 reactions. PN occurred in patients treated with PADCEV with or without preexisting PN. The median time to onset of Grade ≥2 PN was 4.9 months (range: 0.1 to 20 months). Neuropathy led to treatment discontinuation in 6% of patients.
Monitor patients for symptoms of new or worsening PN and consider dose interruption or dose reduction of PADCEV when PN occurs. Permanently discontinue PADCEV in patients who develop Grade ≥3 PN.
Ocular disorders were reported in 40% of the 384 patients treated with PADCEV as a single agent in clinical trials in which ophthalmologic exams were scheduled. The majority of these events involved the cornea and included events associated with dry eye such as keratitis, blurred vision, increased lacrimation, conjunctivitis, limbal stem cell deficiency, and keratopathy. Dry eye symptoms occurred in 30% of patients, and blurred vision occurred in 10% of patients, during treatment with PADCEV. The median time to onset to symptomatic ocular disorder was 1.7 months (range: 0 to 30.6 months). Monitor patients for ocular disorders. Consider artificial tears for prophylaxis of dry eyes and ophthalmologic evaluation if ocular symptoms occur or do not resolve. Consider treatment with ophthalmic topical steroids, if indicated after an ophthalmic exam. Consider dose interruption or dose reduction of PADCEV for symptomatic ocular disorders.
Infusion site extravasation Skin and soft tissue reactions secondary to extravasation have been observed after administration of PADCEV. Of the 720 patients treated with PADCEV as a single agent in clinical trials, 1% of patients experienced skin and soft tissue reactions, including 0.3% who experienced Grade 3-4 reactions. Reactions may be delayed. Erythema, swelling, increased temperature, and pain worsened until 2-7 days after extravasation and resolved within 1-4 weeks of peak. Two patients (0.3%) developed extravasation reactions with secondary cellulitis, bullae, or exfoliation. Ensure adequate venous access prior to starting PADCEV and monitor for possible extravasation during administration. If extravasation occurs, stop the infusion and monitor for adverse reactions.
Embryo-fetal toxicity PADCEV can cause fetal harm when administered to a pregnant woman. Advise patients of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during PADCEV treatment and for 2 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with PADCEV and for 4 months after the last dose.
ADVERSE REACTIONS
Most common adverse reactions, including laboratory abnormalities (≥20%) (PADCEV in combination with pembrolizumab)
Increased aspartate aminotransferase (AST), increased creatinine, rash, increased glucose, PN, increased lipase, decreased lymphocytes, increased alanine aminotransferase (ALT), decreased hemoglobin, fatigue, decreased sodium, decreased phosphate, decreased albumin, pruritus, diarrhea, alopecia, decreased weight, decreased appetite, increased urate, decreased neutrophils, decreased potassium, dry eye, nausea, constipation, increased potassium, dysgeusia, urinary tract infection and decreased platelets.
Most common adverse reactions, including laboratory abnormalities (≥20%) (PADCEV monotherapy)
Increased glucose, increased AST, decreased lymphocytes, increased creatinine, rash, fatigue, PN, decreased albumin, decreased hemoglobin, alopecia, decreased appetite, decreased neutrophils, decreased sodium, increased ALT, decreased phosphate, diarrhea, nausea, pruritus, increased urate, dry eye, dysgeusia, constipation, increased lipase, decreased weight, decreased platelets, abdominal pain, dry skin.
EV-302 Study: 440 patients with previously untreated la/mUC (PADCEV in combination with pembrolizumab)
Serious adverse reactions occurred in 50% of patients treated with PADCEV in combination with pembrolizumab. The most common serious adverse reactions (≥2%) were rash (6%), acute kidney injury (5%), pneumonitis/ILD (4.5%), urinary tract infection (3.6%), diarrhea (3.2%), pneumonia (2.3%), pyrexia (2%), and hyperglycemia (2%). Fatal adverse reactions occurred in 3.9% of patients treated with PADCEV in combination with pembrolizumab including acute respiratory failure (0.7%), pneumonia (0.5%), and pneumonitis/ILD (0.2%).
Adverse reactions leading to discontinuation of PADCEV occurred in 35% of patients. The most common adverse reactions (≥2%) leading to discontinuation of PADCEV were PN (15%), rash (4.1%) and pneumonitis/ILD (2.3%). Adverse reactions leading to dose interruption of PADCEV occurred in 73% of patients. The most common adverse reactions (≥2%) leading to dose interruption of PADCEV were PN (22%), rash (16%), COVID-19 (10%), diarrhea (5%), pneumonitis/ILD (4.8%), fatigue (3.9%), hyperglycemia (3.6%), increased ALT (3%) and pruritus (2.5%). Adverse reactions leading to dose reduction of PADCEV occurred in 42% of patients. The most common adverse reactions (≥2%) leading to dose reduction of PADCEV were rash (16%), PN (13%) and fatigue (2.7%).
EV-103 Study: 121 patients with previously untreated la/mUC who were not eligible for cisplatin-containing chemotherapy (PADCEV in combination with pembrolizumab)
Serious adverse reactions occurred in 50% of patients treated with PADCEV in combination with pembrolizumab; the most common (≥2%) were acute kidney injury (7%), urinary tract infection (7%), urosepsis (5%), sepsis (3.3%), pneumonia (3.3%), hematuria (3.3%), pneumonitis/ILD (3.3%), urinary retention (2.5%), diarrhea (2.5%), myasthenia gravis (2.5%), myositis (2.5%), anemia (2.5%), and hypotension (2.5%). Fatal adverse reactions occurred in 5% of patients treated with PADCEV in combination with pembrolizumab, including sepsis (1.6%), bullous dermatitis (0.8%), myasthenia gravis (0.8%), and pneumonitis/ILD (0.8%). Adverse reactions leading to discontinuation of PADCEV occurred in 36% of patients; the most common (≥2%) were PN (20%) and rash (6%). Adverse reactions leading to dose interruption of PADCEV occurred in 69% of patients; the most common (≥2%) were PN (18%), rash (12%), increased lipase (6%), pneumonitis/ILD (6%), diarrhea (4.1%), acute kidney injury (3.3%), increased ALT (3.3%), fatigue (3.3%), neutropenia (3.3%), urinary tract infection (3.3%), increased amylase (2.5%), anemia (2.5%), COVID-19 (2.5%), hyperglycemia (2.5%), and hypotension (2.5%). Adverse reactions leading to dose reduction of PADCEV occurred in 45% of patients; the most common (≥2%) were PN (17%), rash (12%), fatigue (5%), neutropenia (5%), and diarrhea (4.1%).
EV-301 Study: 296 patients previously treated with a PD-1/L1 inhibitor and platinum-based chemotherapy (PADCEV monotherapy)
Serious adverse reactions occurred in 47% of patients treated with PADCEV; the most common (≥2%) were urinary tract infection, acute kidney injury (7% each), and pneumonia (5%). Fatal adverse reactions occurred in 3% of patients, including multiorgan dysfunction (1%), hepatic dysfunction, septic shock, hyperglycemia, pneumonitis/ILD, and pelvic abscess (0.3% each). Adverse reactions leading to discontinuation occurred in 17% of patients; the most common (≥2%) were PN (5%) and rash (4%). Adverse reactions leading to dose interruption occurred in 61% of patients; the most common (≥4%) were PN (23%), rash (11%), and fatigue (9%). Adverse reactions leading to dose reduction occurred in 34% of patients; the most common (≥2%) were PN (10%), rash (8%), decreased appetite, and fatigue (3% each).
EV-201, Cohort 2 Study: 89 patients previously treated with a PD-1/L1 inhibitor and not eligible for cisplatin-based chemotherapy (PADCEV monotherapy)
Serious adverse reactions occurred in 39% of patients treated with PADCEV; the most common (≥3%) were pneumonia, sepsis, and diarrhea (5% each). Fatal adverse reactions occurred in 8% of patients, including acute kidney injury (2.2%), metabolic acidosis, sepsis, multiorgan dysfunction, pneumonia, and pneumonitis/ILD (1.1% each). Adverse reactions leading to discontinuation occurred in 20% of patients; the most common (≥2%) was PN (7%). Adverse reactions leading to dose interruption occurred in 60% of patients; the most common (≥3%) were PN (19%), rash (9%), fatigue (8%), diarrhea (5%), increased AST, and hyperglycemia (3% each). Adverse reactions leading to dose reduction occurred in 49% of patients; the most common (≥3%) were PN (19%), rash (11%), and fatigue (7%).
DRUG INTERACTIONS
Effects of other drugs on PADCEV (Dual P-gp and Strong CYP3A4 Inhibitors)
Concomitant use with dual P-gp and strong CYP3A4 inhibitors may increase unconjugated monomethyl auristatin E exposure, which may increase the incidence or severity of PADCEV toxicities. Closely monitor patients for signs of toxicity when PADCEV is given concomitantly with dual P-gp and strong CYP3A4 inhibitors.
SPECIFIC POPULATIONS
Lactation Advise lactating women not to breastfeed during treatment with PADCEV and for 3 weeks after the last dose.
Hepatic impairment Avoid the use of PADCEV in patients with moderate or severe hepatic impairment.
Please see full Prescribing Information, including BOXED WARNING.
Important Safety Information/Indication
BOXED WARNING: SERIOUS SKIN REACTIONS
• PADCEV® can cause severe and fatal cutaneous adverse reactions including Stevens-Johnson syndrome (SJS) and Toxic Epidermal Necrolysis (TEN), which occurred predominantly during the first cycle of treatment, but may occur later.
• Closely monitor patients for skin reactions.
• Immediately withhold PADCEV and consider referral for specialized care for suspected SJS or TEN or severe skin reactions.
• Permanently discontinue PADCEV in patients with confirmed SJS or TEN; or Grade 4 or recurrent Grade 3 skin reactions.
INDICATION
PADCEV, in combination with pembrolizumab, is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC).
PADCEV, as a single agent, is indicated for the treatment of adult patients with locally advanced or mUC who:
• have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and platinum-containing chemotherapy, or
• are ineligible for cisplatin-containing chemotherapy and have previously received one or more prior lines of therapy.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Skin reactions Severe cutaneous adverse reactions, including fatal cases of SJS or TEN occurred in patients treated with PADCEV. SJS and TEN occurred predominantly during the first cycle of treatment but may occur later. Skin reactions occurred in 70% (all grades) of the 564 patients treated with PADCEV in combination with pembrolizumab in clinical trials. When PADCEV was given in combination with pembrolizumab, the incidence of skin reactions, including severe events, occurred at a higher rate compared to PADCEV as a single agent. The majority of the skin reactions that occurred with combination therapy included maculo-papular rash, macular rash and papular rash. Grade 3-4 skin reactions occurred in 17% of patients (Grade 3: 16%, Grade 4: 1%), including maculo-papular rash, bullous dermatitis, dermatitis, exfoliative dermatitis, pemphigoid, rash, erythematous rash, macular rash, and papular rash. A fatal reaction of bullous dermatitis occurred in one patient (0.2%). The median time to onset of severe skin reactions was 1.7 months (range: 0.1 to 17.2 months). Skin reactions led to discontinuation of PADCEV in 6% of patients.
Skin reactions occurred in 58% (all grades) of the 720 patients treated with PADCEV as a single agent in clinical trials. Twenty-three percent (23%) of patients had maculo-papular rash and 34% had pruritus. Grade 3-4 skin reactions occurred in 14% of patients, including maculo-papular rash, erythematous rash, rash or drug eruption, symmetrical drug-related intertriginous and flexural exanthema (SDRIFE), bullous dermatitis, exfoliative dermatitis, and palmar-plantar erythrodysesthesia. The median time to onset of severe skin reactions was 0.6 months (range: 0.1 to 8 months). Among patients experiencing a skin reaction leading to dose interruption who then restarted PADCEV (n=75), 24% of patients restarting at the same dose and 24% of patients restarting at a reduced dose experienced recurrent severe skin reactions. Skin reactions led to discontinuation of PADCEV in 3.1% of patients.
Monitor patients closely throughout treatment for skin reactions. Consider topical corticosteroids and antihistamines, as clinically indicated. For persistent or recurrent Grade 2 skin reactions, consider withholding PADCEV until Grade ≤1. Withhold PADCEV and refer for specialized care for suspected SJS, TEN or for Grade 3 skin reactions. Permanently discontinue PADCEV in patients with confirmed SJS or TEN; or Grade 4 or recurrent Grade 3 skin reactions.
Hyperglycemia and diabetic ketoacidosis (DKA), including fatal events, occurred in patients with and without pre-existing diabetes mellitus, treated with PADCEV. Patients with baseline hemoglobin A1C ≥8% were excluded from clinical trials. In clinical trials of PADCEV as a single agent, 17% of the 720 patients treated with PADCEV developed hyperglycemia of any grade; 7% of patients developed Grade 3-4 hyperglycemia (Grade 3: 6.5%, Grade 4: 0.6%). Fatal events of hyperglycemia and DKA occurred in one patient each (0.1%). The incidence of Grade 3-4 hyperglycemia increased consistently in patients with higher body mass index and in patients with higher baseline A1C. The median time to onset of hyperglycemia was 0.5 months (range: 0 to 20 months). Hyperglycemia led to discontinuation of PADCEV in 0.7% of patients. Five percent (5%) of patients required initiation of insulin therapy for treatment of hyperglycemia. Of the patients who initiated insulin therapy for treatment of hyperglycemia, 66% (23/35) discontinued insulin at the time of last evaluation. Closely monitor blood glucose levels in patients with, or at risk for, diabetes mellitus or hyperglycemia. If blood glucose is elevated (>250 mg/dL), withhold PADCEV.
Pneumonitis/Interstitial Lung Disease (ILD) Severe, life-threatening or fatal pneumonitis/ILD occurred in patients treated with PADCEV. When PADCEV was given in combination with pembrolizumab, 10% of the 564 patients treated with combination therapy had pneumonitis/ILD of any grade and 4% had Grade 3-4. A fatal event of pneumonitis/ILD occurred in two patients (0.4%). The incidence of pneumonitis/ILD, including severe events, occurred at a higher rate when PADCEV was given in combination with pembrolizumab compared to PADCEV as a single agent. The median time to onset of any grade pneumonitis/ILD was 4 months (range: 0.3 to 26 months).
In clinical trials of PADCEV as a single agent, 3% of the 720 patients treated with PADCEV had pneumonitis/ILD of any grade and 0.8% had Grade 3-4. The median time to onset of any grade pneumonitis/ILD was 2.9 months (range: 0.6 to 6 months).
Monitor patients for signs and symptoms indicative of pneumonitis/ILD such as hypoxia, cough, dyspnea or interstitial infiltrates on radiologic exams. Evaluate and exclude infectious, neoplastic and other causes for such signs and symptoms through appropriate investigations. Withhold PADCEV for patients who develop Grade 2 pneumonitis/ILD and consider dose reduction. Permanently discontinue PADCEV in all patients with Grade 3 or 4 pneumonitis/ILD.
Peripheral neuropathy (PN) When PADCEV was given in combination with pembrolizumab, 67% of the 564 patients treated with combination therapy had PN of any grade, 36% had Grade 2 neuropathy, and 7% had Grade 3 neuropathy. The incidence of PN occurred at a higher rate when PADCEV was given in combination with pembrolizumab compared to PADCEV as a single agent. The median time to onset of Grade ≥2 PN was 6 months (range: 0.3 to 25 months).
PN occurred in 53% of the 720 patients treated with PADCEV as a single agent in clinical trials including 38% with sensory neuropathy, 8% with muscular weakness and 7% with motor neuropathy. Thirty percent of patients experienced Grade 2 reactions and 5% experienced Grade 3-4 reactions. PN occurred in patients treated with PADCEV with or without preexisting PN. The median time to onset of Grade ≥2 PN was 4.9 months (range: 0.1 to 20 months). Neuropathy led to treatment discontinuation in 6% of patients.
Monitor patients for symptoms of new or worsening PN and consider dose interruption or dose reduction of PADCEV when PN occurs. Permanently discontinue PADCEV in patients who develop Grade ≥3 PN.
Ocular disorders were reported in 40% of the 384 patients treated with PADCEV as a single agent in clinical trials in which ophthalmologic exams were scheduled. The majority of these events involved the cornea and included events associated with dry eye such as keratitis, blurred vision, increased lacrimation, conjunctivitis, limbal stem cell deficiency, and keratopathy. Dry eye symptoms occurred in 30% of patients, and blurred vision occurred in 10% of patients, during treatment with PADCEV. The median time to onset to symptomatic ocular disorder was 1.7 months (range: 0 to 30.6 months). Monitor patients for ocular disorders. Consider artificial tears for prophylaxis of dry eyes and ophthalmologic evaluation if ocular symptoms occur or do not resolve. Consider treatment with ophthalmic topical steroids, if indicated after an ophthalmic exam. Consider dose interruption or dose reduction of PADCEV for symptomatic ocular disorders.
Infusion site extravasation Skin and soft tissue reactions secondary to extravasation have been observed after administration of PADCEV. Of the 720 patients treated with PADCEV as a single agent in clinical trials, 1% of patients experienced skin and soft tissue reactions, including 0.3% who experienced Grade 3-4 reactions. Reactions may be delayed. Erythema, swelling, increased temperature, and pain worsened until 2-7 days after extravasation and resolved within 1-4 weeks of peak. Two patients (0.3%) developed extravasation reactions with secondary cellulitis, bullae, or exfoliation. Ensure adequate venous access prior to starting PADCEV and monitor for possible extravasation during administration. If extravasation occurs, stop the infusion and monitor for adverse reactions.
Embryo-fetal toxicity PADCEV can cause fetal harm when administered to a pregnant woman. Advise patients of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during PADCEV treatment and for 2 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with PADCEV and for 4 months after the last dose.
ADVERSE REACTIONS
Most common adverse reactions, including laboratory abnormalities (≥20%) (PADCEV in combination with pembrolizumab)
Increased aspartate aminotransferase (AST), increased creatinine, rash, increased glucose, PN, increased lipase, decreased lymphocytes, increased alanine aminotransferase (ALT), decreased hemoglobin, fatigue, decreased sodium, decreased phosphate, decreased albumin, pruritus, diarrhea, alopecia, decreased weight, decreased appetite, increased urate, decreased neutrophils, decreased potassium, dry eye, nausea, constipation, increased potassium, dysgeusia, urinary tract infection and decreased platelets.
Most common adverse reactions, including laboratory abnormalities (≥20%) (PADCEV monotherapy)
Increased glucose, increased AST, decreased lymphocytes, increased creatinine, rash, fatigue, PN, decreased albumin, decreased hemoglobin, alopecia, decreased appetite, decreased neutrophils, decreased sodium, increased ALT, decreased phosphate, diarrhea, nausea, pruritus, increased urate, dry eye, dysgeusia, constipation, increased lipase, decreased weight, decreased platelets, abdominal pain, dry skin.
EV-302 Study: 440 patients with previously untreated la/mUC (PADCEV in combination with pembrolizumab)
Serious adverse reactions occurred in 50% of patients treated with PADCEV in combination with pembrolizumab. The most common serious adverse reactions (≥2%) were rash (6%), acute kidney injury (5%), pneumonitis/ILD (4.5%), urinary tract infection (3.6%), diarrhea (3.2%), pneumonia (2.3%), pyrexia (2%), and hyperglycemia (2%). Fatal adverse reactions occurred in 3.9% of patients treated with PADCEV in combination with pembrolizumab including acute respiratory failure (0.7%), pneumonia (0.5%), and pneumonitis/ILD (0.2%).
Adverse reactions leading to discontinuation of PADCEV occurred in 35% of patients. The most common adverse reactions (≥2%) leading to discontinuation of PADCEV were PN (15%), rash (4.1%) and pneumonitis/ILD (2.3%). Adverse reactions leading to dose interruption of PADCEV occurred in 73% of patients. The most common adverse reactions (≥2%) leading to dose interruption of PADCEV were PN (22%), rash (16%), COVID-19 (10%), diarrhea (5%), pneumonitis/ILD (4.8%), fatigue (3.9%), hyperglycemia (3.6%), increased ALT (3%) and pruritus (2.5%). Adverse reactions leading to dose reduction of PADCEV occurred in 42% of patients. The most common adverse reactions (≥2%) leading to dose reduction of PADCEV were rash (16%), PN (13%) and fatigue (2.7%).
EV-103 Study: 121 patients with previously untreated la/mUC who were not eligible for cisplatin-containing chemotherapy (PADCEV in combination with pembrolizumab)
Serious adverse reactions occurred in 50% of patients treated with PADCEV in combination with pembrolizumab; the most common (≥2%) were acute kidney injury (7%), urinary tract infection (7%), urosepsis (5%), sepsis (3.3%), pneumonia (3.3%), hematuria (3.3%), pneumonitis/ILD (3.3%), urinary retention (2.5%), diarrhea (2.5%), myasthenia gravis (2.5%), myositis (2.5%), anemia (2.5%), and hypotension (2.5%). Fatal adverse reactions occurred in 5% of patients treated with PADCEV in combination with pembrolizumab, including sepsis (1.6%), bullous dermatitis (0.8%), myasthenia gravis (0.8%), and pneumonitis/ILD (0.8%). Adverse reactions leading to discontinuation of PADCEV occurred in 36% of patients; the most common (≥2%) were PN (20%) and rash (6%). Adverse reactions leading to dose interruption of PADCEV occurred in 69% of patients; the most common (≥2%) were PN (18%), rash (12%), increased lipase (6%), pneumonitis/ILD (6%), diarrhea (4.1%), acute kidney injury (3.3%), increased ALT (3.3%), fatigue (3.3%), neutropenia (3.3%), urinary tract infection (3.3%), increased amylase (2.5%), anemia (2.5%), COVID-19 (2.5%), hyperglycemia (2.5%), and hypotension (2.5%). Adverse reactions leading to dose reduction of PADCEV occurred in 45% of patients; the most common (≥2%) were PN (17%), rash (12%), fatigue (5%), neutropenia (5%), and diarrhea (4.1%).
EV-301 Study: 296 patients previously treated with a PD-1/L1 inhibitor and platinum-based chemotherapy (PADCEV monotherapy)
Serious adverse reactions occurred in 47% of patients treated with PADCEV; the most common (≥2%) were urinary tract infection, acute kidney injury (7% each), and pneumonia (5%). Fatal adverse reactions occurred in 3% of patients, including multiorgan dysfunction (1%), hepatic dysfunction, septic shock, hyperglycemia, pneumonitis/ILD, and pelvic abscess (0.3% each). Adverse reactions leading to discontinuation occurred in 17% of patients; the most common (≥2%) were PN (5%) and rash (4%). Adverse reactions leading to dose interruption occurred in 61% of patients; the most common (≥4%) were PN (23%), rash (11%), and fatigue (9%). Adverse reactions leading to dose reduction occurred in 34% of patients; the most common (≥2%) were PN (10%), rash (8%), decreased appetite, and fatigue (3% each).
EV-201, Cohort 2 Study: 89 patients previously treated with a PD-1/L1 inhibitor and not eligible for cisplatin-based chemotherapy (PADCEV monotherapy)
Serious adverse reactions occurred in 39% of patients treated with PADCEV; the most common (≥3%) were pneumonia, sepsis, and diarrhea (5% each). Fatal adverse reactions occurred in 8% of patients, including acute kidney injury (2.2%), metabolic acidosis, sepsis, multiorgan dysfunction, pneumonia, and pneumonitis/ILD (1.1% each). Adverse reactions leading to discontinuation occurred in 20% of patients; the most common (≥2%) was PN (7%). Adverse reactions leading to dose interruption occurred in 60% of patients; the most common (≥3%) were PN (19%), rash (9%), fatigue (8%), diarrhea (5%), increased AST, and hyperglycemia (3% each). Adverse reactions leading to dose reduction occurred in 49% of patients; the most common (≥3%) were PN (19%), rash (11%), and fatigue (7%).
DRUG INTERACTIONS
Effects of other drugs on PADCEV (Dual P-gp and Strong CYP3A4 Inhibitors)
Concomitant use with dual P-gp and strong CYP3A4 inhibitors may increase unconjugated monomethyl auristatin E exposure, which may increase the incidence or severity of PADCEV toxicities. Closely monitor patients for signs of toxicity when PADCEV is given concomitantly with dual P-gp and strong CYP3A4 inhibitors.
SPECIFIC POPULATIONS
Lactation Advise lactating women not to breastfeed during treatment with PADCEV and for 3 weeks after the last dose.
Hepatic impairment Avoid the use of PADCEV in patients with moderate or severe hepatic impairment.
Please see full Prescribing Information, including BOXED WARNING.